1/ Presentation of the tutorial
This tutorial will help you install PackMem and guide you through the whole process of packing defect quantification on a test system DOPC/DOPE (70:30). A bash script "ScriptPackMem.sh" which recapitulates most of the steps described in this page is also available.
The main steps are:
- Installation of PackMem
- Extraction of pdb files from an MD trajectory
- PackMem execution on each pdb file
- Histogram analysis and calculation of packing defect constants
- Watch and interpret results
- Add a new lipid in the database
List of dependancies
We recommend to run PackMem on Unix-based OS. To complete this tutorial, you need the following programms to be installed:
- git (download) : under Ubuntu/Debian, a simple "sudo apt-get install git" should do the trick.
- python 2 or 3 (download): already within all recent Linux distributions.
- R (download): under Ubuntu/Debian, a simple "sudo apt-get install r-base-core" should do the trick.
- GROMACS 5.0.7 (download and install, other versions are possible): GROMACS installation is optional, it is used in this tutorial to extract the pdb files from a trajectory in xtc format (GROMACS binary format).
The shell script "ScriptPackMem.sh" has been successfully tested on Ubuntu 16.04.
Before starting the tutorial, we suggest the user to create a new directory in which we will install PackMem, download the test trajectory, and do the complete PackMem analysis:
mkdir test_PackMem
cd test_PackMem
Throughout this tutorial, we also suggest to set a prefix for naming the different files:
prefix="membrane"
Here we set a prefix with a generic name "membrane", but we usually recommand to use the lipid composition (e.g. prefix="dopc70dope30").
2/ PackMem installation
You can skip this section if PackMem is already installed on your computer.
The best way for installing PackMem is to clone the PackMem git repository:
git clone https://github.com/rogautier/packmem.git
You should have now a sub-directory called "packmem" in which all the files are installed. The main program is "packmem/PackMem.py". You can try that everything works by typing:
python "packmem/PackMem.py"
This should ouput the PackMem usage:
usage: PackMem.py [-h] [-i FILENAME] [-r FILESRAD] [-o OUTPUTNAME]
[-d DIST_SUPPL_Z] [-t PD_TYPE] [-p PARAMFILE]
[-n INDEXFILE] [-v]
Command line: python PackMem.py -i file.pdb -r fileRadius.txt -p param.txt -o output -d distGlyc -t deep/all/shallow [-n index file] [-v] or -h for help
If you don't have git installed, you can alternatively download PackMem and unzip the zip file yourself. If you proceed like that be sure to have PackMem installed in a directory called "packmem" if you want to go on with this tutorial.
3/ Extraction of pdb files
We now download and extract an MD traj example from the PackMem website:
wget http://packmem.ipmc.cnrs.fr/download/ExplePackMem.tar.gz
tar -zxvf ExplePackMem.tar.gz
You should have the GROMACS files in the dir called "ExplePackMem".
Create a directory "pdb" to store the trajectory as a collection of pdb files:
mkdir pdb
We first source GROMACS. Then we extract pdb files from the MD trajectory (in xtc format) using the GROMACS tool "trjconv" (one frame every 1000 ps in this case).
We want lipids only (group 9 contains all lipids DOPC and DOPE).
source /usr/local/gromacs-5.0.7/bin/GMXRC
echo 9 | trjconv -f ExplePackMem/dopcdope_exple.xtc \
-s ExplePackMem/dopcdope.tpr -o pdb/${prefix}.pdb -sep \
-n ExplePackMem/index_dopcdope.ndx -dt 100
Now you should have in the "pdb" directory ${prefix}1.pdb, ${prefix}2.pdb, ..., ${prefix}400.pdb.
If you have generated your trajectory using another MD engine, please put all the pdb files of the trajectory you want to analyze, and name them ${prefix}1.pdb, ${prefix}2.pdb, etc.
4/ Launch PackMem on each pdb file
We now want to launch PackMem on each snapshot, we provide for that a bash script called "ScriptPackMem.sh", that you can alternatively get with the Unix tool "wget":
wget http://packmem.ipmc.cnrs.fr/download/ScriptPackMem.sh
Launch the script:
bash ./ScriptPackMem.sh >& OUT &
It'll take a while (a few hours depending on your hardware), if you want to know what pdb file is under analysis, type:
tail -f OUT
When the run is finished, if everything went fine (no python error in "OUT"), you can trash this file:
\rm OUT
5/ Compute packing defects statistics from PackMem results
If the preceding step went well, you should have these three files (each one contains all statistics for each type of packing defect):
- "Total_${prefix}_deep.txt"
- "Total_${prefix}_shallow.txt"
- "Total_${prefix}_all.txt"
Then, you can compute the packing defect distributions using an R script called "Script_fit_and_plot.R". First get it using wget:
wget http://packmem.ipmc.cnrs.fr/download/Script_fit_and_plot.R
And then launch R:
R --vanilla < Script_fit_and_plot.R
The script should create two files:
- Res_membrane.txt: contains all numerical values for each packing defect constant
- Res_membrane.pdf: there are 5 pages which contain:
- p1-3: the distributions for each type of packing defect (deep, shallow, all)
- p4: A barplot showing the packing defect constants with five bars (1: using all data, 2: using a block corresponding to the first third of the data, 3: a block with the second third, 4: a block with the third third, 5: the average of the 3 blocks +/- error (std dev of the three blocks).
- p5: A final barplot containing the packing defect constants +/- error evaluated by block averaging (same as bar 5 of p4).
6/ Watch and interpret results
You can use any pdf viewer to admire your results:
acroread Res_membrane.pdf
You can go to the section Best Practices if you want to have some hints on how to interpret those barplots.
Next to the pdf, a file called "Res_membrane.txt" should contain the following (you can ignore the "[1]" as well as the double-quotes:
[1] "Results on membrane for deep defects"
[1] "Total number of defects = 28238"
[1] "Using all data: 8.7 A^2"
[1] "Using block 1: 15.09 A^2"
[1] "Using block 2: 15.11 A^2"
[1] "Using block 3: 10.04 A^2"
[1] "Mean +/- sd on 3 blocks: 13.42 +/- 2.92 A^2"
[1] ""
[1] "Results on membrane for shallow defects"
[1] "Total number of defects = 42204"
[1] "Using all data: 12.48 A^2"
[1] "Using block 1: 14.2 A^2"
[1] "Using block 2: 13.08 A^2"
[1] "Using block 3: 14.09 A^2"
[1] "Mean +/- sd on 3 blocks: 13.79 +/- 0.61 A^2"
[1] ""
[1] "Results on membrane for all defects"
[1] "Total number of defects = 44376"
[1] "Using all data: 15.76 A^2"
[1] "Using block 1: 18.14 A^2"
[1] "Using block 2: 19.94 A^2"
[1] "Using block 3: 15.68 A^2"
[1] "Mean +/- sd on 3 blocks: 17.92 +/- 2.14 A^2"
[1] ""
This output is useful because it tells you the total number for each type of packing defect. Clearly, the number of defects is not large enough (for the three types of packing defects), which translates into large errors. We provide an archive with more statistics (the useful files are called "TotalCharmm_dopcdope_shallow.txt", "TotalCharmm_dopcdope_deep.txt", "TotalCharmm_dopcdope_all.txt", from a trajectory of 300 ns sampled every 100 ps) on which you can try to run the R script. See also the section Best Practices on which we show a comparison between the undersampled (400 frames) and the fully sampled (3000 frames) examples.
7/ Add a new lipid in the PackMem database
Imagine you have performed a simulation with a lipid that does not exist in the PackMem database. We explain here all the steps needed to add this new lipid in PackMem.
Parameter file
Lipid name
The first part maps the name of the lipid in the PDB file to the lipid name in the program. In PDB format, residue names are only 3-letter long. It can thus lead to issues such as a POPE with the same name as a POPC (i.e. POP). To avoid this, in the script we used the first 2 letters and the final one such as POPC becomes POC and POPE, POE.
To declare a new lipid, do as follows :
POP_H1 POE
Following this convention, if your lipid is declared in the param but does not have the same name in the PDB, put its name in the PDB file together with the first atom name encountered in the lipid.
Example : DOP_H1 DOE
DOE_H1 DOE Glycerol atom
The research of packing defects and the distinction between shallow and deep is dependent on a specific glycerol atom and the -d option (supplied by the user). For phospholipids, we consider the central glycerol atom. To declare it in the parameter file, the first column is the internal name of the lipid (like DOE) followed by the glycerol atom name :
DOE C23
van der Waals file
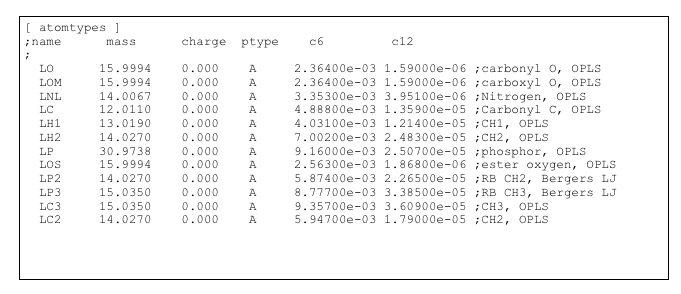
To assess the packing defects, we use the van der Waals radius from the force-field files. In this file we list all atoms of the lipid together with their radius and a letter to identify if the atom is considered aliphatic (a) or not (n). We recommend using the minima of the Lennard-Jones function (corresponding to the Rmin distance) for the radii. The radius is 1/2 of the Rmin with Rmin = 21/6*sigma (sigma can be found in the itp file that declare all the atom types of the forcefield); for GROMOS sigma has to be converted from C6 and C12.
To avoid problems, we advise to use a set of parameter files that are distinct for each force-field. We provide parameters for common lipids in the united-atom Berger and for the all-atom CHARMM forcefields.
Example on DPPC
DPPC in Berger force field:
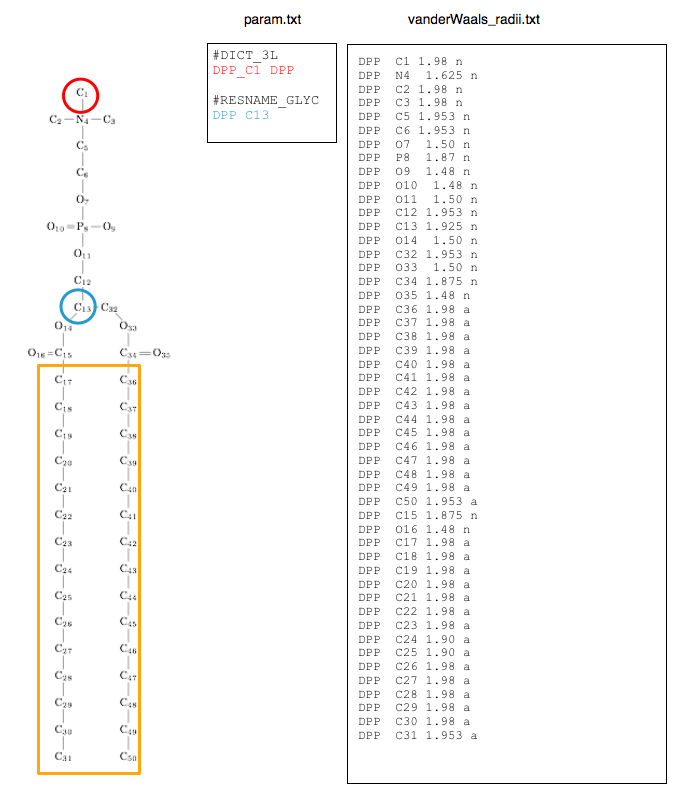
With DPPC.itp : itp from Peter Tieleman website (http://wcm.ucalgary.ca/tieleman/downloads)

And the forcefield file (here lipid.itp): from Peter Tieleman website (http://wcm.ucalgary.ca/tieleman/downloads) where sigma = (C12/C6)1/6